|
MAY 2009 -
J. Am. Chem. Soc. , 2009, 131 (13) , 4892-4903.
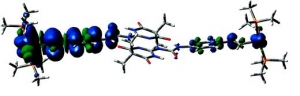
Markus Pichlmaier, Rainer F. Winter, Manfred Zabel and Stanislav Záliš: Electron Transfer Across Multiple Hydrogen Bonds: The Case of Ureapyrimidinedione-Substituted Vinyl Ruthenium and Osmium Complexes, (article here).
Abstract: Ruthenium and osmium complexes 2a,b and 3a,b featuring the N-4,6-dioxo-5,5-dibutyl- or the N-4,6-dioxo-5,5-di-(2-propenyl)-1,4,5,6-tetrahydropyrimidin-2-yl-N′(4-ethenylphenyl)-urea ligand dimerize by a self-complementary quadruply hydrogen-bonding donor/donor/acceptor/acceptor (DDAA) motif. We provide evidence that the dimeric structures are maintained in nonpolar solvents and in 0.1 M NBu4PF6/CH2Cl2 supporting electrolyte solution. All complexes are reversibly oxidized in two consecutive two-electron oxidations (ΔE1/2 ≈ 500 mV) without any discernible potential splitting for the oxidation of the individual hydrogen-bridged redox active moieties. IR and UV/vis/NIR spectroelectrochemistry show a one-step conversion of the neutral to the dication without any discernible features of an intermediate monooxidized radical cation. Oxidation-induced IR changes of the NH and CO groups that are involved in hydrogen bonding are restricted to the styryl-bonded urea NH function. IR band assignments are aided by quantum chemical calculations. Our experimental findings clearly show that, at least in the present systems, the ureapyrimidinedione (Upy) DDAA hydrogen-bonding motif does not support electron transfer. The apparent reason is that neither of the hydrogen-bonding functionalities contributes to the occupied frontier levels. This results in nearly degenerate pairs of MOs representing the in-phase and out-of-phase combinations of the individual monomeric building blocks.
ARCHIVE......
APRIL 2009 -
J. Am. Chem. Soc. , 2009, 131 (12) , 4529-4534.
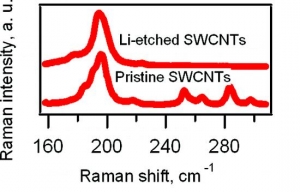
M. Kalbac, L. Kavan, L. Dunsch: Selective Etching of Thin Single Wall Carbon Nanotubes (article here).
Abstract: Raman spectroscopy and in situ Raman spectroelectrochemistry were applied to study the selective etching of thin tubes by lithium vapor in doped single-walled carbon nanotubes (SWCNTs). A strong doping of SWCNTs after the reaction with Li vapor was confirmed by the vanishing of the radial breathing mode (RBM) and by a strong attenuation of the tangential displacement (TG) band in the Raman spectra. The Raman spectra of the Li-vapor-treated SWCNTs after subsequent reaction with water showed changes in the diameter distribution compared with that of a pristine sample (nanotubes with diameters of <1 nm disappeared from the Raman spectra). The samples were tested by the Raman pattern with five different laser lines, and a removal of narrower tubes was confirmed. The remaining wider tubes were not significantly damaged by the treatment with Li, as indicated by the D line in the Raman spectra. Furthermore, the small-diameter tubes are converted not into amorphous carbon but into lithium carbide, which could easily be removed by hydrolysis. The treated samples were further charged electrochemically. It was shown by spectroelectrochemistry that anodic charging may lead to removal of the residual chemical doping from the thicker nanotubes in the sample, but the thin nanotubes did not appear in the spectra. This is a further confirmation of the removal of the small-diameter tubes.
MARCH 2009 -
Analytical chemistry, 2009, 81 (5), 2017-2021.
Kavan L., Janda P., Krause M., et al. : Rotating Cell for in Situ Raman Spectroelectrochemical Studies of Photosensitive Redox Systems (article here).
Abstract: A recently developed rotating spectroelectrochemical. cell for in situ Raman spectroscopic studies of photoreactive compounds without marked decomposition of the sample is presented. Photochemically and thermally sensitive redox systems are difficult to be studied under stationary conditions by in situ spectroelectrochemistry using laser excitation as in Raman spectroscopy. A rotating spectroelectrochemical cell can circumvent these difficulties. It can be used for any type of a planar electrode and for all electrode materials in contact with aqueous or nonaqueous solutions as well as with ionic liquids. The innovative technical solution consists of the precession movement of the spectroelectrochemical cell using an eccentric drive. This precession movement allows a fixed electrical connection to be applied for interfacing the electrochemical cell to a potentiostat. Hence, any electrical imperfections and noise, which would be produced by sliding contacts, are removed. A further advantage of the rotating cell is a dramatic decrease of the thermal load of the electrochemical system. The size of the spectroelectrochemical cell is variable and dependent on the thickness of the cuvettes used ranging up to similar to 10 mm. The larger measuring area causes a higher sensitivity in the spectroscopic studies. The as constructed spectroelectrochemical cell is easy to handle. The performance of the cell is demonstrated for ordered fullerene C-60 layers and the spectroelectrochemical behavior of nanostructured fullerenes. Here the charge transfer at highly ordered fullerene C-60 films was studied by in situ Raman spectroelectrochemistry under appropriate laser power and accumulation time without marked photodecomposition of the sample.
FEBRUARY 2009 -
International Journal of Mass Spectrometry, 2009, 280 (1-3), Pages 1-3.
The Zdenek Herman Honor Issue of the International Journal of Mass Spectrometry appeared in February.
The carrier of Prof. Herman is outlined in the article “Zdenek Herman - An Ambassador of Science” from Veronica M. Bierbaum (University of Colorado, US).

JANUARY 2009 -
Journal of the American Chemical Society, 2009, 131 (2), 494-501
A.Jesenská-J.Sýkora-A.Olzýnska-J.Brezovský-Z.Zdrá́hal-J.Damborský-M.Hof: Nanosecond Time-Dependent Stokes Shift at the Tunnel Mouth of Haloalkane Dehalogenases. (article here).
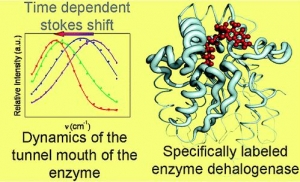
Abstract: The tunnel mouths are evolutionally the most variable regions in the structures of haloalkane dehalogenases originating from different bacterial species, suggesting their importance for adaptation of enzymes to various substrates. We decided to monitor the dynamics of this particular region by means of time-resolved fluorescence spectroscopy and molecular dynamic simulations. To label the enzyme specifically, we adapted a novel procedure that utilizes a coumarin dye containing a halide−hydrocarbon linker, which serves as a substrate for enzymatic reaction. The procedure leads to a coumarin dye covalently attached and specifically located in the tunnel mouth of the enzyme. In this manner, we stained two haloalkane dehalogenase mutants, DbjA-H280F and DhaA-H272F. The measurements of time-resolved fluorescence anisotropy, acrylamide quenching, and time-resolved emission spectra reveal differences in the polarity, accessibility and mobility of the dye and its microenvironment for both of the mutants. The obtained experimental data are consistent with the results obtained by molecular dynamics calculations and correlate with the anatomy of the tunnel mouths, which were proposed to have a strong impact on the catalytic activity and specificity of the examined mutants. Interestingly, the kinetics of the recorded time-dependent Stokes shift is unusual slow; it occurs on the nanosecond time-scale, suggesting that the protein dynamics is extremely slowed down at the region involved in the exchange of ligands between the active-site cavity and bulk solvent.
|
 Struktura
Struktura 


