|
OCTOBER 2009 -
ACS Nano, 2009, 3 (8), pp. 2320-2328.
Martin Kalbac, Hootan Farhat, Ladislav Kavan, Jing Kong, Ken-ichi Sasaki, Riichiro
Saito and Mildred S. Dresselhaus: Electrochemical Charging of Individual Single-Walled Carbon Nanotubes (article here).
Abstract:
The influence of the electrode potential on the electronic structure of individual single-walled carbon nanotubes is studied using Raman spectroscopy. By analyzing the radial breathing mode intensity versus electrode potential profiles in the Raman spectra at many different laser excitation energies, we show that the charging of individual carbon nanotubes causes a broadening of the resonant Raman profiles (resonance window). This effect is observed for both a semiconducting and a metallic tube. The broadening of the resonance Raman profiles already begins at potentials where the first electronic states of a particular tube are filled or depleted. The important consequence of this effect is a striking difference between the Raman intensity versus potential profiles of metallic and semiconducting tubes. While for a metallic tube the intensity of the Raman signal is attenuated at potentials which deviate slightly from 0 V, for a semiconducting tube, the Raman intensity is significantly attenuated only after the electrode potential reaches the first van Hove singularity. Furthermore, for the metallic tube, a strong asymmetry is found in the bleaching of the Raman signal with respect to positive and negative potentials, which results from the different energy bandwidth for the π* band and the π band.
J. Am. Chem. Soc. , 2009, 131 (33), pp 11788–11800.
Ana Mara Blanco-Rodrguez, Michael Busby, Kate Ronayne, Michael Towrie, Cristian Grdinaru, Jawahar Sudhamsu, Jan Sýkora, Martin Hof, Stanislav Záliš, Angel J. Di Bilio, Brian R. Crane, Harry B. Gray and Antonín Vlček, Jr.: Relaxation Dynamics of Pseudomonas aeruginosa ReI(CO)3(α-diimine)(HisX)+ (X = 83, 107, 109, 124, 126)CuII Azurins (article here).
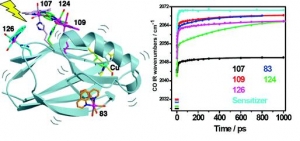
Abstract: Photoinduced relaxation processes of five structurally characterized Pseudomonas aeruginosa ReI(CO)3(α-diimine)(HisX) (X = 83, 107, 109, 124, 126)CuII azurins have been investigated by time-resolved (ps−ns) IR spectroscopy and emission spectroscopy. Crystal structures reveal the presence of Re-azurin dimers and trimers that in two cases (X = 107, 124) involve van der Waals interactions between interdigitated diimine aromatic rings. Time-dependent emission anisotropy measurements confirm that the proteins aggregate in mM solutions (D2O, KPi buffer, pD = 7.1). Excited-state DFT calculations show that extensive charge redistribution in the ReI(CO)3 → diimine 3MLCT state occurs: excitation of this 3MLCT state triggers several relaxation processes in Re-azurins whose kinetics strongly depend on the location of the metallolabel on the protein surface. Relaxation is manifested by dynamic blue shifts of excited-state ν(CO) IR bands that occur with triexponential kinetics: intramolecular vibrational redistribution together with vibrational and solvent relaxation give rise to subps, 2, and 8−20 ps components, while the 102 ps kinetics are attributed to displacement (reorientation) of the ReI(CO)3(phen)(im) unit relative to the peptide chain, which optimizes Coulombic interactions of the ReI excited-state electron density with solvated peptide groups. Evidence also suggests that additional segmental movements of Re-bearing β-strands occur without perturbing the reaction field or interactions with the peptide. Our work demonstrates that time-resolved IR spectroscopy and emission anisotropy of ReI carbonyl−diimine complexes are powerful probes of molecular dynamics at or around the surfaces of proteins and protein−protein interfacial regions.
ARCHIVE......
SEPTEMBER 2009 -
Analytical Chemistry, 2009, 81, 6382-6389.
Langmaier J., Samec Z.: Voltammetry of Ion Transfer across a Polarized Room-Temperature Ionic Liquid Membrane Facilitated by Valinomycin: Theoretical Aspects and Application (article here).
Abstract: Cyclic voltammetry is used to investigate the transfer of alkali-metal cations, protons, and ammonium ions facilitated by the complex formation with valinomycin at the interface between an aqueous electrolyte solution and a room-temperature ionic liquid (RTIL) membrane. The membrane is made of a thin (112 μm) microporous filter impregnated with an RTIL that is composed of tridodecylmethylammonium cations and tetrakis[3,5-bis(trifluoromethyl)phenyl]borate anions. An extension of the existing theory of voltammetry of ion transfer across polarized liquid membranes makes it possible to evaluate the standard ion-transfer potentials for the hydrophilic cations studied, as well as the stability constants (Ki) of their 1:1 complexes with valinomycin, as log Ki = 9.0 (H+), 11.1 (Li+), 12.8 (Na+), 17.2 (K+), 15.7 (Rb+), 15.1 (Cs+), and 14.7 (NH4+). These data point to the remarkably enhanced stability of the valinomycin complexes within RTIL, and to the enhanced selectivity of valinomycin for K+ over all other univalent ions studied, compared to the conventional K+ ion-selective liquid-membrane electrodes. Selective complex formation allows one to resolve voltammetric responses of K+ and Na+ in the presence of an excess of Mg2+ or Ca2+, which is demonstrated by determination of K+ and Na+ in the table and tap water samples.
Analytical Chemistry, 2009, 81, 6327-6333.
Mikysek T., Švancara I., Klacher K., Bartoš M., Vytřas K. Ludvík J.: New Approaches to the Characterization of Carbon Paste Electrodes Using the Ohmic Resistance Effect and Qualitative Carbon Paste Indexes (article here).
Abstract: In this article, some new approaches to characterize the carbon paste mixtures and the respective carbon paste electrodes (CPEs) are presented, discussed, and critically evaluated. Particular attention has been paid to the changes of the ohmic resistance, relative to the dependence on composition of the CPE, the materials used, the time, and the position of storage. Four types of carbon pastes were examined, and for the interpretation of experimental data, a new simple model of “close-packing of spheres” has been applied. This model resembles the percolation theory for solid matter. In our case, however, it is possible to explain not only the “bent” or “broken” shape of the dependence of the electrode resistance upon the binder:carbon ratio and the corresponding electrochemical current response, but also differences caused by various material used and three various effects observed during the electrode aging. Furthermore, the report presents the significance of practical utilization of the recently introduced carbon paste index (denoted as χCPE), which is a qualitative hitherto unused factor based on the evaluation of cyclic voltammograms for standard redox systems (e.g., [Fe(CN)6]3−/4−) and specifying the electrochemical properties of a CPE. Some problems connected with homogeneity and stability of carbon pastes, their handling, storage, or eventual aging effects are also discussed.
AUGUST 2009 -
Biophysical Journal, 2009, 97 (3), L1-L3.
Štefl M., Kułakowska A., Hof M.: Simultaneous Characterization of Lateral Lipid and Prothrombin Diffusion Coefficients by Z-Scan Fluorescence Correlation Spectroscopy (article here).
Abstract: A new (to our knowledge) robust approach for the determination of lateral diffusion coefficients of weakly bound proteins is applied for the phosphatidylserine specific membrane interaction of bovine prothrombin. It is shown that z-scan fluorescence correlation spectroscopy in combination with pulsed interleaved dual excitation allows simultaneous monitoring of the lateral diffusion of labeled protein and phospholipids. Moreover, from the dependencies of the particle numbers on the axial sample positions at different protein concentrations phosphatidylserine-dependent equilibrium dissociation constants are derived confirming literature values. Increasing the amount of membrane-bound prothrombin retards the lateral protein and lipid diffusion, indicating coupling of both processes. The lateral diffusion coefficients of labeled lipids are considerably larger than the simultaneously determined lateral diffusion coefficients of prothrombin, which contradicts findings reported for the isolated N-terminus of prothrombin.
Journal of Catalysis, 2009, 266, 79-91.
Zilkova N., Bejblova M., Gil B., Zones S.I., Burton A. W., Chen C. Y., Musilova-Pavlackova Z., Kosova G., Cejka J.: The role of the zeolite channel architecture and acidity on the activity and selectivity in aromatic transformations: The effect of zeolite cages in SSZ-35 zeolite (article here).
Abstract: A series of zeolites differing in the channel architecture and acidity was investigated in toluene disproportionation, together with toluene and p-xylene alkylation with isopropyl alcohol. Zeolites with one- to three-dimensional 10-ring and 12-ring channels with and without cages, and those having 12–12–10 and 12–10–10-ring channel systems were studied. It was shown that general relationship of increasing zeolite activity with increasing pore diameter and pore connectivity is not valid as the size of some 12-ring channels (Beta, MCM-68) is comparable with 10-ring channels (ZSM-5, SSZ-35). In addition, the presence of cages in the structure of SSZ-35 and MCM-58 attributes the unusual catalytic behavior of these zeolites. SSZ-35 and MCM-58 zeolites behave in both toluene reactions such as three-dimensional large-pore zeolites. Subtle differences between zeolites of similar pore sizes and dimensionality can be usually explained based on the differences in the acidity of the individual zeolites. In p-xylene alkylation SSZ-35 exhibited high conversion with the highest selectivity to 1-isopropyl-2,5-dimethyl benzene and a low rate of deactivation. The presence of 18-ring cages in the channels of 10-ring zeolite SSZ-35 (STF) gives rise to an unusual catalytic behavior of this zeolite by comparison to other 10-ring zeolites. SSZ-35, possessing channels of 0.54 × 0.57 nm in diameter, exhibits catalytic activity in transformation of aromatic hydrocarbons that is similar to large-pore
zeolites. 18-Ring cages enable the formation of relatively bulkier transition states while the diffusion of the product molecules out of the 10-ring channel system is not slowed down due to 10-ring windows. In addition, the channel system of SSZ-35 prevents the formation of coke precursors.
JULY 2009 -
Journal of Catalysis, 2009, 262 (1), 27-34.
Jisa K., Novakova J., Schwarze M., Vondrova A., Sklenak S., Sobalik Z.: Role of the Fe-zeolite structure and iron state in the N2O decomposition: Comparison of Fe-FER, Fe-BEA, and Fe-MFI catalysts (article here).
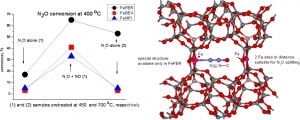
Abstract: The decomposition of nitrous oxide was compared over Fe-FER, Fe-MFI, and Fe-BEA with well established iron distribution in cationic positions and low amounts of less well-established oxide species. It was evidenced that, despite a comparable content of Fe(II) in the cationic positions, the catalytic activity of re-FER greatly exceeds that of Fe-BEA and Fe-MFI. While about one half of the iron sites in Fe-FER (Fe/Al < 0.15) participate in the decomposition of nitrous oxide after activation at 450 degrees C, the number of active sites in Fe-BEA or Fe-MFI was much lower, and, accordingly, without acceleration of the reaction by the addition of NO, these samples exhibit much lower catalytic activity than Fe-FER. This could be likely correlated with the concentration of Fe(II) in positions with a specific spatial iron arrangement at optimal Fe center dot center dot center dot Fe distances. For that role we propose a local structure with two adjacent beta sites, where the Fe center dot center dot center dot Fe distance would be 7 to 7.5 angstrom, i.e. comparable to the length of the N2O molecule, and provide potential for cooperation of the two iron cations on the N2O Molecule splitting. Such arrangement is absent in both the Fe-BEA and re-MFI structures.
JUNE 2009 -
Chemistry of Materials, 2009, 21 (8), 1457-1464.
Prochazka J., Kavan L., Zukalova M., Frank O., Kalbac M., Zukal A., Klementova M., Carbone D.,
Graetzel M.: Novel Synthesis of the TiO2(B) Multilayer Templated Films.
Abstract: TiO2(B) mesoporous thin films were grown in two steps on the F-doped SnO2 conductive glass substrates. In the first step, a small amount of H3PO4, corresponding to 0.15-0.375 wt % P on TiO2 basis, was introduced into concentrated HCl which was subsequently used for hydrolysis of titanium ethoxide. The hydrolyzed colloidal TiO2 Suspension was further mixed with a 1-butanol solution of the amphiphilic triblock copolymer Pluronic P123. The obtained precursor mixture was used for dip coating of FTO substrates. To achieve over 1 mu m thick films, dip coating (followed by a thermal treatment at 350 degrees C/2 h) was repeated several times to produce multilayer films. The films consisted of amorphous TiO2 with small amounts of anatase and TiO2(B). The amorphous part was converted into the TiO2(B) in a simple firing step at 500-550 degrees C. The formation of TiO2(B) phase was accompanied by a significant increase of the film thickness. The films demonstrated unique behavior during the electrochemical lithium insertion that would qualify them for fast battery or electrochromic smart window applications. The efficiency of multiphase TiO2 films in dye sensitized solar cells depends on the composition of individual films: it increases in the series: anatase/ amorphous TiO2 < anatase/TiO2(B) < anatase.
MAY 2009 -
J. Am. Chem. Soc. , 2009, 131 (13) , 4892-4903.
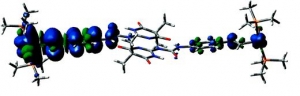
Markus Pichlmaier, Rainer F. Winter, Manfred Zabel and Stanislav Záliš: Electron Transfer Across Multiple Hydrogen Bonds: The Case of Ureapyrimidinedione-Substituted Vinyl Ruthenium and Osmium Complexes, (article here).
Abstract: Ruthenium and osmium complexes 2a,b and 3a,b featuring the N-4,6-dioxo-5,5-dibutyl- or the N-4,6-dioxo-5,5-di-(2-propenyl)-1,4,5,6-tetrahydropyrimidin-2-yl-N′(4-ethenylphenyl)-urea ligand dimerize by a self-complementary quadruply hydrogen-bonding donor/donor/acceptor/acceptor (DDAA) motif. We provide evidence that the dimeric structures are maintained in nonpolar solvents and in 0.1 M NBu4PF6/CH2Cl2 supporting electrolyte solution. All complexes are reversibly oxidized in two consecutive two-electron oxidations (ΔE1/2 ≈ 500 mV) without any discernible potential splitting for the oxidation of the individual hydrogen-bridged redox active moieties. IR and UV/vis/NIR spectroelectrochemistry show a one-step conversion of the neutral to the dication without any discernible features of an intermediate monooxidized radical cation. Oxidation-induced IR changes of the NH and CO groups that are involved in hydrogen bonding are restricted to the styryl-bonded urea NH function. IR band assignments are aided by quantum chemical calculations. Our experimental findings clearly show that, at least in the present systems, the ureapyrimidinedione (Upy) DDAA hydrogen-bonding motif does not support electron transfer. The apparent reason is that neither of the hydrogen-bonding functionalities contributes to the occupied frontier levels. This results in nearly degenerate pairs of MOs representing the in-phase and out-of-phase combinations of the individual monomeric building blocks.
APRIL 2009 -
J. Am. Chem. Soc. , 2009, 131 (12) , 4529-4534.
M. Kalbac, L. Kavan, L. Dunsch: Selective Etching of Thin Single Wall Carbon Nanotubes (article here).
Abstract: Raman spectroscopy and in situ Raman spectroelectrochemistry were applied to study the selective etching of thin tubes by lithium vapor in doped single-walled carbon nanotubes (SWCNTs). A strong doping of SWCNTs after the reaction with Li vapor was confirmed by the vanishing of the radial breathing mode (RBM) and by a strong attenuation of the tangential displacement (TG) band in the Raman spectra. The Raman spectra of the Li-vapor-treated SWCNTs after subsequent reaction with water showed changes in the diameter distribution compared with that of a pristine sample (nanotubes with diameters of <1 nm disappeared from the Raman spectra). The samples were tested by the Raman pattern with five different laser lines, and a removal of narrower tubes was confirmed. The remaining wider tubes were not significantly damaged by the treatment with Li, as indicated by the D line in the Raman spectra. Furthermore, the small-diameter tubes are converted not into amorphous carbon but into lithium carbide, which could easily be removed by hydrolysis. The treated samples were further charged electrochemically. It was shown by spectroelectrochemistry that anodic charging may lead to removal of the residual chemical doping from the thicker nanotubes in the sample, but the thin nanotubes did not appear in the spectra. This is a further confirmation of the removal of the small-diameter tubes.
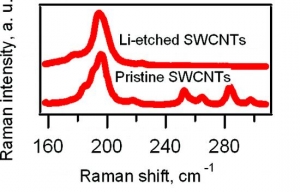
MARCH 2009 -
Analytical chemistry, 2009, 81 (5), 2017-2021.
Kavan L., Janda P., Krause M., et al. : Rotating Cell for in Situ Raman Spectroelectrochemical Studies of Photosensitive Redox Systems (article here).
Abstract: A recently developed rotating spectroelectrochemical. cell for in situ Raman spectroscopic studies of photoreactive compounds without marked decomposition of the sample is presented. Photochemically and thermally sensitive redox systems are difficult to be studied under stationary conditions by in situ spectroelectrochemistry using laser excitation as in Raman spectroscopy. A rotating spectroelectrochemical cell can circumvent these difficulties. It can be used for any type of a planar electrode and for all electrode materials in contact with aqueous or nonaqueous solutions as well as with ionic liquids. The innovative technical solution consists of the precession movement of the spectroelectrochemical cell using an eccentric drive. This precession movement allows a fixed electrical connection to be applied for interfacing the electrochemical cell to a potentiostat. Hence, any electrical imperfections and noise, which would be produced by sliding contacts, are removed. A further advantage of the rotating cell is a dramatic decrease of the thermal load of the electrochemical system. The size of the spectroelectrochemical cell is variable and dependent on the thickness of the cuvettes used ranging up to similar to 10 mm. The larger measuring area causes a higher sensitivity in the spectroscopic studies. The as constructed spectroelectrochemical cell is easy to handle. The performance of the cell is demonstrated for ordered fullerene C-60 layers and the spectroelectrochemical behavior of nanostructured fullerenes. Here the charge transfer at highly ordered fullerene C-60 films was studied by in situ Raman spectroelectrochemistry under appropriate laser power and accumulation time without marked photodecomposition of the sample.
FEBRUARY 2009 -
International Journal of Mass Spectrometry, 2009, 280 (1-3), Pages 1-3.
The Zdenek Herman Honor Issue of the International Journal of Mass Spectrometry appeared in February.
The carrier of Prof. Herman is outlined in the article “Zdenek Herman - An Ambassador of Science” from Veronica M. Bierbaum (University of Colorado, US).

JANUARY 2009 -
Journal of the American Chemical Society, 2009, 131 (2), 494-501
A.Jesenská-J.Sýkora-A.Olzýnska-J.Brezovský-Z.Zdrá́hal-J.Damborský-M.Hof: Nanosecond Time-Dependent Stokes Shift at the Tunnel Mouth of Haloalkane Dehalogenases. (article here).
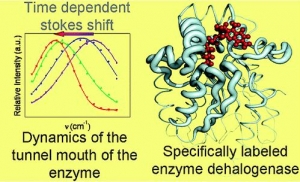
Abstract: The tunnel mouths are evolutionally the most variable regions in the structures of haloalkane dehalogenases originating from different bacterial species, suggesting their importance for adaptation of enzymes to various substrates. We decided to monitor the dynamics of this particular region by means of time-resolved fluorescence spectroscopy and molecular dynamic simulations. To label the enzyme specifically, we adapted a novel procedure that utilizes a coumarin dye containing a halide−hydrocarbon linker, which serves as a substrate for enzymatic reaction. The procedure leads to a coumarin dye covalently attached and specifically located in the tunnel mouth of the enzyme. In this manner, we stained two haloalkane dehalogenase mutants, DbjA-H280F and DhaA-H272F. The measurements of time-resolved fluorescence anisotropy, acrylamide quenching, and time-resolved emission spectra reveal differences in the polarity, accessibility and mobility of the dye and its microenvironment for both of the mutants. The obtained experimental data are consistent with the results obtained by molecular dynamics calculations and correlate with the anatomy of the tunnel mouths, which were proposed to have a strong impact on the catalytic activity and specificity of the examined mutants. Interestingly, the kinetics of the recorded time-dependent Stokes shift is unusual slow; it occurs on the nanosecond time-scale, suggesting that the protein dynamics is extremely slowed down at the region involved in the exchange of ligands between the active-site cavity and bulk solvent.
|





