|
JULY 2010 -
Angew. Chem. Int. Ed. 2010, 49, 4813-4815.
V. Petrykin, K. Macounova, O. A. Shlyakhtin, P. Krtil: Tailoring the Selectivity for Electrocatalytic Oxygen Evolution on Ruthenium Oxides by Zinc Substitution (article here).
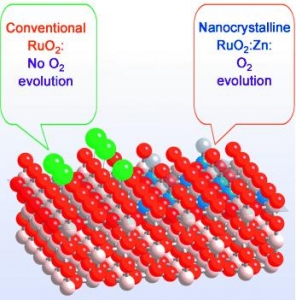
Abstract: Controlling gas emissions: Versatile control of the selectivity of an oxide electrocatalyst in the oxygen- and chlorine-evolution reactions was demonstrated by Zn substitution in RuO2 (see picture: O red, Cl green, Zn blue, Ru white). The incorporation of Zn into the rutile structure alters the cation sequence along the [001] direction and modifies the structure of the active sites for both gas-evolution processes.
Chem. Eur. J. 2010, 16, 7773 – 7780.
Dana Procházková, Martina Bejblová, Josef Vlk, Ajayan Vinu, Petr Štěpnička, Jiří Čejka: Selective Monoacylation of Ferrocene with Bulky Acylating Agents over Mesoporous Sieve AlKIT-5 (article here).
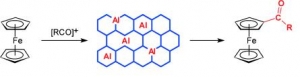
Abstract: Ferrocene acylation with bulky acylating agents (1-adamantoyl, benzoyl, 2-chlorobenzoyl, and cinnamoyl chlorides; and benzoic anhydride) catalyzed by AlKIT-5 mesoporous catalysts was investigated. AlKIT-5 catalysts with varying ratios of Si/Al were synthesized using tetraethoxysilane and aluminum isopropoxide as the structural building blocks and Pluronic F127 as a template under acidic conditions and were characterized in detail by X-ray powder diffraction, magic-angle spinning (MAS) NMR spectroscopy, sorption of nitrogen, energy-dispersive X-ray spectroscopy (EDS), SEM, TEM, and FTIR with pyridine as a probe molecule. The catalytic activity of the prepared AlKIT-5 catalysts in ferrocene acylation was shown to depend on the type of the acylating agent, thus likely reflecting the strength of interactions between the acyl source, the product, and the solid catalysts when the acylation reaction was carried out at 100 °C. In all reactions, the AlKIT-5 catalysts afforded exclusively the monoacylated products (100 % selectivity) most likely due to deactivation of the second cyclopentadiene ring by attachment of the first acyl group, steric reasons, and some competitive interactions of the monoacylferrocenes with the catalysts. The prepared AlKIT-5 catalysts could be regenerated without any significant loss of the ferrocene conversion.
ARCHIVE......
JUNE 2010 -
Coordination Chemistry Reviews, 2010, 254, 1383–1396.
S. Záliš, R.F. Winter, W. Kaim: Quantum chemical interpretation of redox properties of ruthenium complexes with vinyl and TCNX type non-innocent ligands (article here).
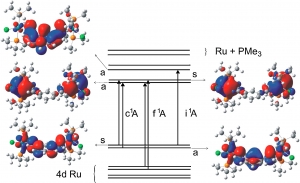
Abstract: This review provides an overview of density functional theory (DFT) calculations in a consequence with spectroelectrochemical measurements on mononuclear and symmetrically or unsymmetrically bridged di- and tetranuclear ruthenium complexes of vinyl and TCNX ligands. The DFT approach is used for the calculations of molecular structures, vibrational frequencies, electronic and electron paramagnetic resonance (EPR) spectral data. DFT calculations enable us to identity the primary redox site and the electron and spin-density distribution between the individual components for the individual redox congeners. The DFT technique reproduces the spectral properties of the presented complexes and their radical ions. The generally close correspondence between experimental and quantum chemical results demonstrate that modern DFT is a powerful tool to address issues like ligand non-innocence and electron and spin delocalization in systems containing both redox- ctive metal ions and redox-active ligands.
Journal of Catalysis, 2010, 272 (2), 262-274.
Š. Sklenák, P. C. Andrikopoulos, B. Boekfa, B. Jansang, J. Nováková, L. Benco, T. Bucko, J. Hafner, J. Dědeček, Z. Sobalík: N2O decomposition over Fe-zeolites: Structure of the active sites and the origin of the distinct reactivity of Fe-ferrierite, Fe-ZSM-5, and Fe-beta. A combined periodic DFT and multispectral study (article here).
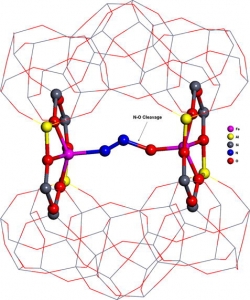
Abstract: The N2O decomposition over Fe-ferrierite, Fe-beta, and Fe-ZSM-5 has been recently studied [K. Jisa, J. Novakova, M. Schwarze, A. Vondrova, S. Sklenak, Z. Sobalik, J. Catal. 262 (2009) 27] and a superior activity of Fe-ferrierite with respect to Fe-beta and Fe-ZSM-5 has been shown. In this study, we investigated (1) plausible active sites for the N2O decomposition over Fe-ferrierite and (2) the origin of the distinct reactivity of Fe-ferrierite, Fe-ZSM-5 and Fe-beta employing a combined theoretical (periodic DFT) and experimental (UV–vis–NIR spectroscopy, IR spectroscopy, 29Si MAS NMR spectroscopy and catalytic batch experiments) approach. We evidenced that two Fe(II) cations accommodated in two adjacent six-membered rings in the eight-membered ring channel (β sites) of Fe-ferrierite (the calculated Fe–Fe distance is 7.4 Å) form the active site responsible for the superior activity of this catalyst in the N2O decomposition in the absence of NO. Similar structures can be formed in Fe-beta. However, the probability of their formation is very low. For Fe-ZSM-5, the geometrical arrangement of the cationic positions is far from that in Fe-ferrierite and it is not suitable for the N2O decomposition. Therefore, the predicted order of the activity of the Fe(II) exchanged zeolites agrees with our experimental findings and it is: Fe-ferrierite much greater-than Fe-beta > Fe-ZSM-5. We further showed that the accommodation of divalent cations in rings forming cationic sites can lead to significant rearrangements of the local structures of the zeolite framework, and therefore, the precise structure of sites binding a divalent cation cannot be derived from results of X-ray diffraction experiments, but can be inferred from theoretical calculations.
MAY 2010 -
BBA - Biomembranes, Vol.1798, 7, 2010, 1377-1391.
Radek Macháň, Martin Hof: Lipid diffusion in planar membranes investigated by fluorescence correlation spectroscopy (artile here).
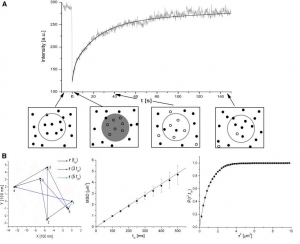
Abstract: Investigation of lipid lateral mobility in biological membranes and their artificial models provides information on membrane dynamics and structure; methods based on optical microscopy are very convenient for such investigations. We focus on fluorescence correlation spectroscopy (FCS), explain its principles and review its state of the art versions such as 2-focus, Z-scan or scanning FCS, which overcome most artefacts of standard FCS (especially those resulting from the need for an external calibration) making it a reliable and versatile method. FCS is also compared to single particle tracking and fluorescence photobleaching recovery and the applicability and the limitations of the methods are briefly reviewed. We discuss several key questions of lateral mobility investigation in planar lipid membranes, namely the influence which membrane and aqueous phase composition (ionic strength and sugar content), choice of a fluorescent tracer molecule, frictional coupling between the two membrane leaflets and between membrane and solid support
(in the case of supported membranes) or presence of membrane inhomogeneities has on the lateral mobility of lipids. The recent FCS studies addressing those questions are reviewed and possible explanations of eventual discrepancies are mentioned.
Chem. Mater., 2010, 22 (11), pp 3482–3495.
Oleksiy V. Shvets, Natalia Kasian, Arnošt Zukal, Jiří Pinkas, and Jiří Čejka: The Role of Template Structure and Synergism between Inorganic and Organic Structure Directing Agents in the Synthesis of UTL Zeolite (article here).
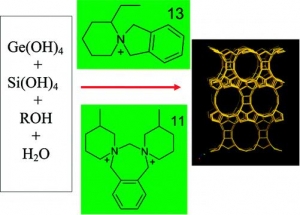
Abstract: The extra-large-pore germanosilicates with UTL topology have been synthesized using a large variety of spiroazocompounds as structure-directing agents. Synthesis conditions were optimized and zeolites with a high crystallinity degree were obtained with 13 different organic structure-directing agents. The influence of the composition of the reaction mixture and template nature (structure, hydrophilicity/hydro-phobicity balance, rigidity, pKa) on the phase selectivity, crystallinity degree, and adsorption properties of zeolites with UTL structure was investigated. Selection criteria of organic molecules as potential structure-directing agents (SDAs) in the synthesis of large-pore and extra-large-pore zeolites from silicate and germanosilicate media are proposed. The optimum synthesis time was determined to be 4−9 days for different SDA and (Si + Ge)/SDA molar ratios. Clear synergism between the optimum structure of organic template and the presence of critical amount of inorganic component (GeO2) was evidenced. The UTL zeolite crystallizes as tiny sheets 10 μm thick. The effect of the organic template on the size and shape of the crystals was found. The micropore volume of the best crystals is 0.22−0.24 cm3/g, with a micropore diameter of 1.05 nm, based on density functional theory (DFT), and Saito−Foley analyses of adsorption isotherms.
APRIL 2010 -
Phys. Chem. Chem. Phys., 2010, 12, 5240-5247.
Arnošt Zukal, Jana Mayerová and Jiří Čejka: Alkali metal cation doped Al-SBA-15 for carbon dioxide adsorption (article here).
Abstract: Mesoporous aluminosilicate adsorbents for carbon dioxide were prepared by the grafting of aluminium into SBA-15 silica using an aqueous solution of aluminium chlorohydrate. As the ion exchange sites are primarily associated with the presence of tetrahedrally coordinated aluminium, extra-framework aluminium on the SBA-15 surface was inserted into the silica matrix by a treatment with an aqueous solution of NH4OH. Synthesized mesoporous aluminosilicate preserving all the characteristic features of a mesoporous molecular sieve was finally modified by the alkali metal cation exchange. To examine carbon dioxide adsorption on prepared materials, adsorption isotherms in the temperature range from 0 °C to 60 °C were measured. Based on the known temperature dependence of adsorption isotherms, isosteric adsorption heats giving information on the surface energetics of CO2 adsorption were calculated and discussed. The comparison of carbon dioxide isotherms obtained on aluminosilicate SBA-15, aluminosilicate SBA-15 containing cations Na+ and K+ and activated alumina F-200 reveals that the doping with sodium or potassium cations dramatically enhances adsorption in the region of equilibrium pressures lower than 10 kPa. Therefore, synthesized aluminosilicate adsorbents doped with Na+ or K+ cations are suitable for carbon dioxide separation from dilute gas mixtures.
Zeolites and Catalysis: Synthesis, Reactions and Applications
Jiří Čejka (Editor), Avelino Corma (Editor), Stacey Zones (Editor). Wiley, ISBN:
978-3-527-32514-6, 918 pages, April 2010.

This indispensable two-volume handbook covers everything on this hot research field.
The first part deals with the synthesis, modification, characterization and application of catalytic active zeolites, while the second focuses on such reaction types as cracking, hydrocracking, isomerization, reforming and other industrially important topics. Edited by a highly experienced and internationally renowned team with chapters written by the "Who's Who" of zeolite research (more info about book...).
MARCH 2010 -
Angew. Chem. Int. Ed. 2010, 49, 2937 –2940.
Martin Lamač, Anke Spannenberg, Haijun Jiao, Sven Hansen, Wolfgang Baumann, Perdita Arndt, Uwe Rosenthal: Formation of a 1-Zircona-2,5-disilacyclopent-3-yne: Coordination of 1,4-Disilabutatriene to Zirconocene? (article here)
Abstract: Alkyne under stress: A novel metallacycle containing one Zr atom, two Si
atoms, and a CC bond has been prepared and its structure elucidated. According to X-ray data, spectral properties, and DFT calculations, the bonding situation in this compound is characterized as a 1-metalla-2,5-disilacyclopent-3-yne with a weak metal-triple-bond interaction.
Phys. Chem. Chem. Phys., 2010, 12 (13), 3145-3155.
Votava O., Mašát M., Pracna P., Kassi S., Campargue A.: Accurate determination of low state rotational quantum numbers (J < 4) from planar-jet and liquid nitrogen cell absorption spectra of methane near 1.4 micron (article here).
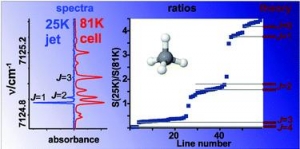
Abstract: An improved procedure for accurate determination of empirical lower state rotational quantum numbers from molecular absorption spectra is demonstrated for methane. We combine the high resolution absorption spectra in the 7070-7300 cm(-1) frequency range obtained in liquid nitrogen cooled cryogenic cell (T = 81 K) and in supersonic planar jet expansion (T-R = 25 K). Empirical lower state energies of 59 transitions are determined from the ratio of the absolute absorption line strengths at 25 and 81 K. The procedure relies on the realistic description of rotational state populations in the supersonic jet expansion where non-equilibrium nuclear spin isomer distributions are generated due to the rapid cooling. The accuracy of the experimental determination of the lower state energies with J <= 3 is found to considerably improve the results of the same approach applied to spectra at 296 and 81 K. The 59 transitions with determined lower J values provide a good starting point for the theoretical interpretation of the highly congested icosad region of methane. In particular, the centres of nine vibrational bands are estimated from the transitions with J = 0 lower state rotational quantum number.
FEBRUARY 2010 -
J.Am.Chem. Soc., 2010,132(8), 2655–2662.
Bin Su, Imren Hatay, Antonín Trojánek, Zdeněk Samec, Tony Khoury, Claude P. Gros, Jean-Michel Barbe, Antoine Daina, Pierre-Alain Carrupt and Hubert H. Girault: Molecular Electrocatalysis for Oxygen Reduction by Cobalt Porphyrins Adsorbed at Liquid/Liquid Interfaces (article here).
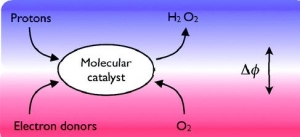
Abstract: Molecular electrocatalysis for oxygen reduction at a polarized water/1,2- dichloroethane (DCE) interface was studied, involving aqueous protons, ferrocene (Fc) in DCE and amphiphilic cobalt porphyrin catalysts adsorbed at the interface. The catalyst, (2,8,13,17-tetraethyl-3,7,12,18-tetramethyl-5-p-amino-phenylporphyrin) cobalt(II) (CoAP), functions like conventional cobalt porphyrins, activating O2 via coordination by the formation of a superoxide structure. Furthermore, due to the hydrophilic nature of the aminophenyl group, CoAP has a strong affinity for the water/DCE interface as evidenced by lipophilicity mapping calculations and surface tension measurements, facilitating the protonation of the CoAP−O2 complex and its reduction by ferrocene. The reaction is electrocatalytic as its rate depends on the applied Galvani potential difference between the two phases.
JANUARY 2010 -
Phys. Chem. Chem. Phys., 20100, 12, 1550-1556.
Lubomír Pospíšil, Filip Teplý, Miroslav Gál, Louis Adriaenssens, Michal Horáček and Lukáš Severa: Helquats, helical extended diquats, as fast electron transfer systems (article here).
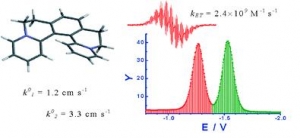
Abstract:
Helicene-viologen structural hybrids, like [5]helquat, 6,7,10,11-tetrahydrodipyrido[2,1-a:1´,2´-k][2,9]phenanthrolinediium, and its four methylated derivatives, are characterized by electrochemical admittance and EPR spectroscopy. All compounds are reversibly reduced in two one-electron steps. Formal redox potentials correlate with the calculated LUMO energies. The electron transfer is coupled with a weak adsorption of the reactants. The analysis of the frequency dependence of the electrode admittance is used for the separation of Faradaic and double layer contributions and finally to the estimation of heterogeneous rate constants. Heterogeneous rate constants determined this way are in the range 0.1 to 3 cm s-1. In all cases the second electron transfer is faster than the first redox step by a factor of three. The Frumkin correction for the acceleration by the double layer potential further amplifies this difference. The heterogeneous rate constants of derivatives correlate with the solvent reorganization energy estimated from the Marcus model. EPR spectra confirm the radical cation formation. The radical of [5]helquat participates in an extremely fast self-exchange process with the parent dication characterized by the self-exchange rate constant kET = (2.4 ± 0.5) × 109 M-1 s-1.
ACS Nano, 2010, 4 (1), pp. 459-469.
Martin Kalbáč, Alexander A. Green, Mark C. Hersam, and Ladislav Kavan: Tuning of Sorted Double-Walled Carbon Nanotubes by Electrochemical Charging (article here).

Abstract: Double-walled carbon nanotubes sorted by density gradient ltracentrifugation were examined by Raman spectroscopy and by in situ Raman pectroelectrochemistry. The sorted samples had a narrow distribution of diameters of oth inner and outer tubes, which enabled a comparison of the behavior of inner metallic tubes and inner semiconducting nanotubes as a function of the applied electrochemical potential. The metallic inner tubes were efficiently doped even though they were protected from electrolyte ions by the outer wall, whereas the doping of emiconducting inner tubes was observed only at high magnitudes of the electrode otential. These results indicate that the doping response of inner tubes is redominantly controlled by inner tube electronic properties. On the other hand, the effect of electronic structure of the outer tube on the behavior of inner tube is weak. Furthermore, the fficiency of the charge transfer from outer to inner wall depends on the doping level. A low doping level corresponds to a high efficiency of the charge-transfer, while a high doping level shows low charge-transfer efficiency.
DECEMBER 2009 -
CARBON, 2009, 48 (1), 153-162.
Pospisil L, Hromadova M, Gal M, et al.: Redox potentials and binding enhancement of fullerene and fullerene-cyclodextrin systems in water and dimethylsulfoxide (article here).
Abstract: The formal redox potentials of electron transfer reactions of fullerene, methanofullerene, ullerene–cyclodextrin complex and methanofullerene conjugates with cyclodextrins in aqueous and imethylsulfoxide solutions are reported. These new compounds are surface active and retain the redox activity of C60 even in aqueous medium. Compounds have been characterized by an electrochemical dmittance technique, which offers an advantage of separating faradaic and capacitive properties. Observed difference of formal redox potentials of the free fullerene forms and their cyclodextrin- ontaining compounds were used to determine the binding enhancement. Results are interpreted in terms of inter-molecular host–guest interactions of C60-cyclodextrin conjugates.
NOVEMBER 2009 -
Biophysical Journal, 2009, 97 ( 9), 2623-2629.
Jana Humpolíčková, Aleš Benda and Jörg Enderlein: Optical Saturation as a Versatile Tool to Enhance Resolution in Confocal Microscopy (article here).
Abstract:
One of the most actively developing areas in fluorescence microscopy is the achievement of spatial resolution below Abbe's diffraction limit, which restricts the resolution to several hundreds of nanometers. Most of the approaches in use at this time require a complex optical setup, a difficult mathematical treatment, or usage of dyes with special photophysical properties. In this work, we present a new, to our knowledge, approach in confocal microscopy that enhances the resolution moderately but is both technically and computationally simple. As it is based on the saturation of the transition from the ground state to the first excited state, it is universally applicable with respect to the dye used. The idea of the method presented is based on a principle similar to that underlying saturation excitation microscopy, but instead of applying harmonically modulated excitation light, the fluorophores are excited by picosecond laser pulses at different intensities, resulting in different levels of saturation. We show that the method can be easily combined with the concept of triplet relaxation, which by tuning the dark periods between pulses helps to suppress the formation of a photolabile triplet state and effectively reduces photobleaching. We demonstrate our approach imaging GFP-labeled protein patches within the plasma membrane of yeast cells.
Chem. Soc. Rev., 2009, 38, 3373-3382.
W. Kaim and J. Fiedler: Spectroelectrochemistry: the best of two worlds (article here).
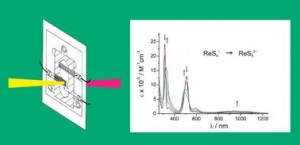
Abstract:
The combination of reaction-oriented electrochemistry with species-focussed spectroscopy in spectroelectrochemistry (SEC) allows for a more complete analysis of single and multiple electron-transfer processes and redox reactions in general. Practical considerations and guidelines are provided in this tutorial review, and selected examples involving UV-VIS-NIR and IR absorption spectroscopy as well as electron paramagnetic resonance (EPR) are presented to illustrate the potential and the applicability of this technique.
OCTOBER 2009 -
ACS Nano, 2009, 3 (8), pp. 2320-2328.
Martin Kalbac, Hootan Farhat, Ladislav Kavan, Jing Kong, Ken-ichi Sasaki, Riichiro
Saito and Mildred S. Dresselhaus: Electrochemical Charging of Individual Single-Walled Carbon Nanotubes (article here).
Abstract:
The influence of the electrode potential on the electronic structure of individual single-walled carbon nanotubes is studied using Raman spectroscopy. By analyzing the radial breathing mode intensity versus electrode potential profiles in the Raman spectra at many different laser excitation energies, we show that the charging of individual carbon nanotubes causes a broadening of the resonant Raman profiles (resonance window). This effect is observed for both a semiconducting and a metallic tube. The broadening of the resonance Raman profiles already begins at potentials where the first electronic states of a particular tube are filled or depleted. The important consequence of this effect is a striking difference between the Raman intensity versus potential profiles of metallic and semiconducting tubes. While for a metallic tube the intensity of the Raman signal is attenuated at potentials which deviate slightly from 0 V, for a semiconducting tube, the Raman intensity is significantly attenuated only after the electrode potential reaches the first van Hove singularity. Furthermore, for the metallic tube, a strong asymmetry is found in the bleaching of the Raman signal with respect to positive and negative potentials, which results from the different energy bandwidth for the π* band and the π band.
J. Am. Chem. Soc. , 2009, 131 (33), pp 11788–11800.
Ana Mara Blanco-Rodrguez, Michael Busby, Kate Ronayne, Michael Towrie, Cristian Grdinaru, Jawahar Sudhamsu, Jan Sýkora, Martin Hof, Stanislav Záliš, Angel J. Di Bilio, Brian R. Crane, Harry B. Gray and Antonín Vlček, Jr.: Relaxation Dynamics of Pseudomonas aeruginosa ReI(CO)3(α-diimine)(HisX)+ (X = 83, 107, 109, 124, 126)CuII Azurins (article here).
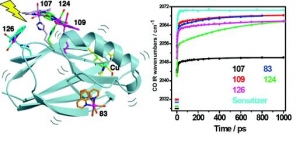
Abstract: Photoinduced relaxation processes of five structurally characterized Pseudomonas aeruginosa ReI(CO)3(α-diimine)(HisX) (X = 83, 107, 109, 124, 126)CuII azurins have been investigated by time-resolved (ps−ns) IR spectroscopy and emission spectroscopy. Crystal structures reveal the presence of Re-azurin dimers and trimers that in two cases (X = 107, 124) involve van der Waals interactions between interdigitated diimine aromatic rings. Time-dependent emission anisotropy measurements confirm that the proteins aggregate in mM solutions (D2O, KPi buffer, pD = 7.1). Excited-state DFT calculations show that extensive charge redistribution in the ReI(CO)3 → diimine 3MLCT state occurs: excitation of this 3MLCT state triggers several relaxation processes in Re-azurins whose kinetics strongly depend on the location of the metallolabel on the protein surface. Relaxation is manifested by dynamic blue shifts of excited-state ν(CO) IR bands that occur with triexponential kinetics: intramolecular vibrational redistribution together with vibrational and solvent relaxation give rise to subps, 2, and 8−20 ps components, while the 102 ps kinetics are attributed to displacement (reorientation) of the ReI(CO)3(phen)(im) unit relative to the peptide chain, which optimizes Coulombic interactions of the ReI excited-state electron density with solvated peptide groups. Evidence also suggests that additional segmental movements of Re-bearing β-strands occur without perturbing the reaction field or interactions with the peptide. Our work demonstrates that time-resolved IR spectroscopy and emission anisotropy of ReI carbonyl−diimine complexes are powerful probes of molecular dynamics at or around the surfaces of proteins and protein−protein interfacial regions.
SEPTEMBER 2009 -
Analytical Chemistry, 2009, 81, 6382-6389.
Langmaier J., Samec Z.: Voltammetry of Ion Transfer across a Polarized Room-Temperature Ionic Liquid Membrane Facilitated by Valinomycin: Theoretical Aspects and Application (article here).
Abstract: Cyclic voltammetry is used to investigate the transfer of alkali-metal cations, protons, and ammonium ions facilitated by the complex formation with valinomycin at the interface between an aqueous electrolyte solution and a room-temperature ionic liquid (RTIL) membrane. The membrane is made of a thin (112 μm) microporous filter impregnated with an RTIL that is composed of tridodecylmethylammonium cations and tetrakis[3,5-bis(trifluoromethyl)phenyl]borate anions. An extension of the existing theory of voltammetry of ion transfer across polarized liquid membranes makes it possible to evaluate the standard ion-transfer potentials for the hydrophilic cations studied, as well as the stability constants (Ki) of their 1:1 complexes with valinomycin, as log Ki = 9.0 (H+), 11.1 (Li+), 12.8 (Na+), 17.2 (K+), 15.7 (Rb+), 15.1 (Cs+), and 14.7 (NH4+). These data point to the remarkably enhanced stability of the valinomycin complexes within RTIL, and to the enhanced selectivity of valinomycin for K+ over all other univalent ions studied, compared to the conventional K+ ion-selective liquid-membrane electrodes. Selective complex formation allows one to resolve voltammetric responses of K+ and Na+ in the presence of an excess of Mg2+ or Ca2+, which is demonstrated by determination of K+ and Na+ in the table and tap water samples.
Analytical Chemistry, 2009, 81, 6327-6333.
Mikysek T., Švancara I., Klacher K., Bartoš M., Vytřas K. Ludvík J.: New Approaches to the Characterization of Carbon Paste Electrodes Using the Ohmic Resistance Effect and Qualitative Carbon Paste Indexes (article here).
Abstract: In this article, some new approaches to characterize the carbon paste mixtures and the respective carbon paste electrodes (CPEs) are presented, discussed, and critically evaluated. Particular attention has been paid to the changes of the ohmic resistance, relative to the dependence on composition of the CPE, the materials used, the time, and the position of storage. Four types of carbon pastes were examined, and for the interpretation of experimental data, a new simple model of “close-packing of spheres” has been applied. This model resembles the percolation theory for solid matter. In our case, however, it is possible to explain not only the “bent” or “broken” shape of the dependence of the electrode resistance upon the binder:carbon ratio and the corresponding electrochemical current response, but also differences caused by various material used and three various effects observed during the electrode aging. Furthermore, the report presents the significance of practical utilization of the recently introduced carbon paste index (denoted as χCPE), which is a qualitative hitherto unused factor based on the evaluation of cyclic voltammograms for standard redox systems (e.g., [Fe(CN)6]3−/4−) and specifying the electrochemical properties of a CPE. Some problems connected with homogeneity and stability of carbon pastes, their handling, storage, or eventual aging effects are also discussed.
AUGUST 2009 -
Biophysical Journal, 2009, 97 (3), L1-L3.
Štefl M., Kułakowska A., Hof M.: Simultaneous Characterization of Lateral Lipid and Prothrombin Diffusion Coefficients by Z-Scan Fluorescence Correlation Spectroscopy (article here).
Abstract: A new (to our knowledge) robust approach for the determination of lateral diffusion coefficients of weakly bound proteins is applied for the phosphatidylserine specific membrane interaction of bovine prothrombin. It is shown that z-scan fluorescence correlation spectroscopy in combination with pulsed interleaved dual excitation allows simultaneous monitoring of the lateral diffusion of labeled protein and phospholipids. Moreover, from the dependencies of the particle numbers on the axial sample positions at different protein concentrations phosphatidylserine-dependent equilibrium dissociation constants are derived confirming literature values. Increasing the amount of membrane-bound prothrombin retards the lateral protein and lipid diffusion, indicating coupling of both processes. The lateral diffusion coefficients of labeled lipids are considerably larger than the simultaneously determined lateral diffusion coefficients of prothrombin, which contradicts findings reported for the isolated N-terminus of prothrombin.
Journal of Catalysis, 2009, 266, 79-91.
Zilkova N., Bejblova M., Gil B., Zones S.I., Burton A. W., Chen C. Y., Musilova-Pavlackova Z., Kosova G., Cejka J.: The role of the zeolite channel architecture and acidity on the activity and selectivity in aromatic transformations: The effect of zeolite cages in SSZ-35 zeolite (article here).
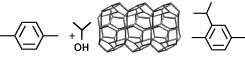
Abstract: A series of zeolites differing in the channel architecture and acidity was investigated in toluene disproportionation, together with toluene and p-xylene alkylation with isopropyl alcohol. Zeolites with one- to three-dimensional 10-ring and 12-ring channels with and without cages, and those having 12–12–10 and 12–10–10-ring channel systems were studied. It was shown that general relationship of increasing zeolite activity with increasing pore diameter and pore connectivity is not valid as the size of some 12-ring channels (Beta, MCM-68) is comparable with 10-ring channels (ZSM-5, SSZ-35). In addition, the presence of cages in the structure of SSZ-35 and MCM-58 attributes the unusual catalytic behavior of these zeolites. SSZ-35 and MCM-58 zeolites behave in both toluene reactions such as three-dimensional large-pore zeolites. Subtle differences between zeolites of similar pore sizes and dimensionality can be usually explained based on the differences in the acidity of the individual zeolites. In p-xylene alkylation SSZ-35 exhibited high conversion with the highest selectivity to 1-isopropyl-2,5-dimethyl benzene and a low rate of deactivation. The presence of 18-ring cages in the channels of 10-ring zeolite SSZ-35 (STF) gives rise to an unusual catalytic behavior of this zeolite by comparison to other 10-ring zeolites. SSZ-35, possessing channels of 0.54 × 0.57 nm in diameter, exhibits catalytic activity in transformation of aromatic hydrocarbons that is similar to large-pore
zeolites. 18-Ring cages enable the formation of relatively bulkier transition states while the diffusion of the product molecules out of the 10-ring channel system is not slowed down due to 10-ring windows. In addition, the channel system of SSZ-35 prevents the formation of coke precursors.
JULY 2009 -
Journal of Catalysis, 2009, 262 (1), 27-34.
Jisa K., Novakova J., Schwarze M., Vondrova A., Sklenak S., Sobalik Z.: Role of the Fe-zeolite structure and iron state in the N2O decomposition: Comparison of Fe-FER, Fe-BEA, and Fe-MFI catalysts (article here).
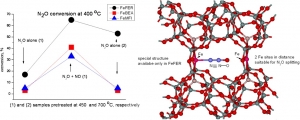
Abstract: The decomposition of nitrous oxide was compared over Fe-FER, Fe-MFI, and Fe-BEA with well established iron distribution in cationic positions and low amounts of less well-established oxide species. It was evidenced that, despite a comparable content of Fe(II) in the cationic positions, the catalytic activity of re-FER greatly exceeds that of Fe-BEA and Fe-MFI. While about one half of the iron sites in Fe-FER (Fe/Al < 0.15) participate in the decomposition of nitrous oxide after activation at 450 degrees C, the number of active sites in Fe-BEA or Fe-MFI was much lower, and, accordingly, without acceleration of the reaction by the addition of NO, these samples exhibit much lower catalytic activity than Fe-FER. This could be likely correlated with the concentration of Fe(II) in positions with a specific spatial iron arrangement at optimal Fe center dot center dot center dot Fe distances. For that role we propose a local structure with two adjacent beta sites, where the Fe center dot center dot center dot Fe distance would be 7 to 7.5 angstrom, i.e. comparable to the length of the N2O molecule, and provide potential for cooperation of the two iron cations on the N2O Molecule splitting. Such arrangement is absent in both the Fe-BEA and re-MFI structures.
JUNE 2009 -
Chemistry of Materials, 2009, 21 (8), 1457-1464.
Prochazka J., Kavan L., Zukalova M., Frank O., Kalbac M., Zukal A., Klementova M., Carbone D.,
Graetzel M.: Novel Synthesis of the TiO2(B) Multilayer Templated Films.
Abstract: TiO2(B) mesoporous thin films were grown in two steps on the F-doped SnO2 conductive glass substrates. In the first step, a small amount of H3PO4, corresponding to 0.15-0.375 wt % P on TiO2 basis, was introduced into concentrated HCl which was subsequently used for hydrolysis of titanium ethoxide. The hydrolyzed colloidal TiO2 Suspension was further mixed with a 1-butanol solution of the amphiphilic triblock copolymer Pluronic P123. The obtained precursor mixture was used for dip coating of FTO substrates. To achieve over 1 mu m thick films, dip coating (followed by a thermal treatment at 350 degrees C/2 h) was repeated several times to produce multilayer films. The films consisted of amorphous TiO2 with small amounts of anatase and TiO2(B). The amorphous part was converted into the TiO2(B) in a simple firing step at 500-550 degrees C. The formation of TiO2(B) phase was accompanied by a significant increase of the film thickness. The films demonstrated unique behavior during the electrochemical lithium insertion that would qualify them for fast battery or electrochromic smart window applications. The efficiency of multiphase TiO2 films in dye sensitized solar cells depends on the composition of individual films: it increases in the series: anatase/ amorphous TiO2 < anatase/TiO2(B) < anatase.
MAY 2009 -
J. Am. Chem. Soc. , 2009, 131 (13) , 4892-4903.
Markus Pichlmaier, Rainer F. Winter, Manfred Zabel and Stanislav Záliš: Electron Transfer Across Multiple Hydrogen Bonds: The Case of Ureapyrimidinedione-Substituted Vinyl Ruthenium and Osmium Complexes, (article here).
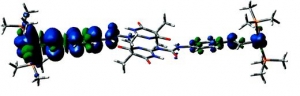
Abstract: Ruthenium and osmium complexes 2a,b and 3a,b featuring the N-4,6-dioxo-5,5-dibutyl- or the N-4,6-dioxo-5,5-di-(2-propenyl)-1,4,5,6-tetrahydropyrimidin-2-yl-N′(4-ethenylphenyl)-urea ligand dimerize by a self-complementary quadruply hydrogen-bonding donor/donor/acceptor/acceptor (DDAA) motif. We provide evidence that the dimeric structures are maintained in nonpolar solvents and in 0.1 M NBu4PF6/CH2Cl2 supporting electrolyte solution. All complexes are reversibly oxidized in two consecutive two-electron oxidations (ΔE1/2 ≈ 500 mV) without any discernible potential splitting for the oxidation of the individual hydrogen-bridged redox active moieties. IR and UV/vis/NIR spectroelectrochemistry show a one-step conversion of the neutral to the dication without any discernible features of an intermediate monooxidized radical cation. Oxidation-induced IR changes of the NH and CO groups that are involved in hydrogen bonding are restricted to the styryl-bonded urea NH function. IR band assignments are aided by quantum chemical calculations. Our experimental findings clearly show that, at least in the present systems, the ureapyrimidinedione (Upy) DDAA hydrogen-bonding motif does not support electron transfer. The apparent reason is that neither of the hydrogen-bonding functionalities contributes to the occupied frontier levels. This results in nearly degenerate pairs of MOs representing the in-phase and out-of-phase combinations of the individual monomeric building blocks.
APRIL 2009 -
J. Am. Chem. Soc. , 2009, 131 (12) , 4529-4534.
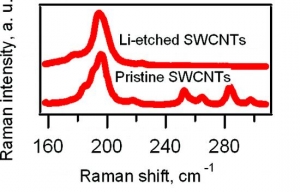
M. Kalbac, L. Kavan, L. Dunsch: Selective Etching of Thin Single Wall Carbon Nanotubes (article here).
Abstract: Raman spectroscopy and in situ Raman spectroelectrochemistry were applied to study the selective etching of thin tubes by lithium vapor in doped single-walled carbon nanotubes (SWCNTs). A strong doping of SWCNTs after the reaction with Li vapor was confirmed by the vanishing of the radial breathing mode (RBM) and by a strong attenuation of the tangential displacement (TG) band in the Raman spectra. The Raman spectra of the Li-vapor-treated SWCNTs after subsequent reaction with water showed changes in the diameter distribution compared with that of a pristine sample (nanotubes with diameters of <1 nm disappeared from the Raman spectra). The samples were tested by the Raman pattern with five different laser lines, and a removal of narrower tubes was confirmed. The remaining wider tubes were not significantly damaged by the treatment with Li, as indicated by the D line in the Raman spectra. Furthermore, the small-diameter tubes are converted not into amorphous carbon but into lithium carbide, which could easily be removed by hydrolysis. The treated samples were further charged electrochemically. It was shown by spectroelectrochemistry that anodic charging may lead to removal of the residual chemical doping from the thicker nanotubes in the sample, but the thin nanotubes did not appear in the spectra. This is a further confirmation of the removal of the small-diameter tubes.
MARCH 2009 -
Analytical chemistry, 2009, 81 (5), 2017-2021.
Kavan L., Janda P., Krause M., et al. : Rotating Cell for in Situ Raman Spectroelectrochemical Studies of Photosensitive Redox Systems (article here).
Abstract: A recently developed rotating spectroelectrochemical. cell for in situ Raman spectroscopic studies of photoreactive compounds without marked decomposition of the sample is presented. Photochemically and thermally sensitive redox systems are difficult to be studied under stationary conditions by in situ spectroelectrochemistry using laser excitation as in Raman spectroscopy. A rotating spectroelectrochemical cell can circumvent these difficulties. It can be used for any type of a planar electrode and for all electrode materials in contact with aqueous or nonaqueous solutions as well as with ionic liquids. The innovative technical solution consists of the precession movement of the spectroelectrochemical cell using an eccentric drive. This precession movement allows a fixed electrical connection to be applied for interfacing the electrochemical cell to a potentiostat. Hence, any electrical imperfections and noise, which would be produced by sliding contacts, are removed. A further advantage of the rotating cell is a dramatic decrease of the thermal load of the electrochemical system. The size of the spectroelectrochemical cell is variable and dependent on the thickness of the cuvettes used ranging up to similar to 10 mm. The larger measuring area causes a higher sensitivity in the spectroscopic studies. The as constructed spectroelectrochemical cell is easy to handle. The performance of the cell is demonstrated for ordered fullerene C-60 layers and the spectroelectrochemical behavior of nanostructured fullerenes. Here the charge transfer at highly ordered fullerene C-60 films was studied by in situ Raman spectroelectrochemistry under appropriate laser power and accumulation time without marked photodecomposition of the sample.
FEBRUARY 2009 -
International Journal of Mass Spectrometry, 2009, 280 (1-3), Pages 1-3.
The Zdenek Herman Honor Issue of the International Journal of Mass Spectrometry appeared in February.
The carrier of Prof. Herman is outlined in the article “Zdenek Herman - An Ambassador of Science” from Veronica M. Bierbaum (University of Colorado, US).

JANUARY 2009 -
Journal of the American Chemical Society, 2009, 131 (2), 494-501
A.Jesenská-J.Sýkora-A.Olzýnska-J.Brezovský-Z.Zdrá́hal-J.Damborský-M.Hof: Nanosecond Time-Dependent Stokes Shift at the Tunnel Mouth of Haloalkane Dehalogenases. (article here).
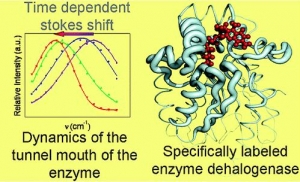
Abstract: The tunnel mouths are evolutionally the most variable regions in the structures of haloalkane dehalogenases originating from different bacterial species, suggesting their importance for adaptation of enzymes to various substrates. We decided to monitor the dynamics of this particular region by means of time-resolved fluorescence spectroscopy and molecular dynamic simulations. To label the enzyme specifically, we adapted a novel procedure that utilizes a coumarin dye containing a halide−hydrocarbon linker, which serves as a substrate for enzymatic reaction. The procedure leads to a coumarin dye covalently attached and specifically located in the tunnel mouth of the enzyme. In this manner, we stained two haloalkane dehalogenase mutants, DbjA-H280F and DhaA-H272F. The measurements of time-resolved fluorescence anisotropy, acrylamide quenching, and time-resolved emission spectra reveal differences in the polarity, accessibility and mobility of the dye and its microenvironment for both of the mutants. The obtained experimental data are consistent with the results obtained by molecular dynamics calculations and correlate with the anatomy of the tunnel mouths, which were proposed to have a strong impact on the catalytic activity and specificity of the examined mutants. Interestingly, the kinetics of the recorded time-dependent Stokes shift is unusual slow; it occurs on the nanosecond time-scale, suggesting that the protein dynamics is extremely slowed down at the region involved in the exchange of ligands between the active-site cavity and bulk solvent.
|
 Struktura
Struktura 



