








Oko, kterým pozorujeme svět kolem nás, je skvěle uzpůsobeno pro detekci záření různých vlnových délek. Světlo procházející čočkou je zaostřeno na oční sítnici (lat. retina), kde se nacházejí desítky milionů buněk, které světlo citlivě vnímají a zprostředkovávají nám přesný a barevný obraz okolního prostředí. Při dědičném onemocnění sítnice zvaném retinitis pigmentosa tyto světločivné buňky odumírají, což vede k postupnému zhoršování zraku a v nejhorších případech až k oslepnutí.
Toto onemocnění, kterým trpí tři až čtyři miliony lidí na celém světě, je způsobeno mutacemi různých genů, z nichž většina hraje důležitou roli v procesu detekce světla, např. genu pro opsin.
K výzkumu mutací způsobujících retinitis pigmentosa jsme se dostali oklikou. Hlavním zájmem Oddělení biologie RNA (Ústav molekulární genetiky AV ČR, v. v. i.) je způsob, jakým se v buňce zpracovává informace uložená v DNA. Konkrétně nás zajímají úpravy na úrovni pre-mRNA, tzv. pre-mRNA sestřih a komplexy, které sestřih katalyzují. Pre-mRNA molekuly jsou přesnou kopií DNA a než mohou sloužit jako templát pro syntézu proteinu, musí z nich být odstraněno více jak 9/10 sekvencí. Zbylé úseky jsou přesně spojeny, aby z nich složená informace byla čitelná a dávala smysl tak, aby mohl být na jejím základě vyroben funkční protein. Tento fascinující proces byl objeven před 30 lety, a i když jsou jeho základní zákonitosti popsány, stále není jasné, jak buňka dokáže přesně a rychle rozpoznat sekvence, které mají být odstraněny a které mají zůstat.
Vědci ve Velké Británii před několika lety zjistili, že dědičnou retinitis pigmentosa způsobují kromě mutací genů specifických pro oko i mutace v genech, jež jsou nezbytné pro přežití každé buňky v našem těle. Největším překvapením pro nás bylo, že většina těchto esenciálních genů kóduje bílkoviny důležité pro pre-mRNA sestřih! Protože jsme tyto sestřihové bílkoviny důvěrně znali a měli dobře zmapováno chování jejich nemutovaných forem, mohli jsme velmi rychle otestovat hypotézu, že mutace v první řadě ovlivňují správné formování sestřihových komplexů.
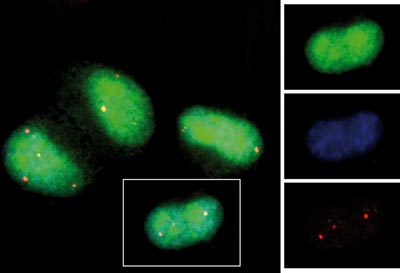
Záchrana buněk exprimujících mutovaný hPrp31 (zelený) pomocí hPrp6 (modrý). Červeně jsou označena nově vzniklá Cajalova tělíska, která indikují zlepšenou formu buněk obsahujících hPrp6.
Foto: Archiv autora
Soustředili jsme se na gen kódující protein hPrp31, který je důležitou součástí sestřihových RNA-proteinových komplexů. Zjistili jsme, že bodová mutace, která způsobuje retinitis pigmentosa, výrazně ovlivňuje chování tohoto proteinu v buňce. Zasažený protein se špatně začleňuje do sestřihových komplexů. Navíc váže – a tím inaktivuje – další bílkovinu, hPrp6, která je pro pre-mRNA důležitá. Dále jsme zjistili, že přítomnost vadného hPrp31 způsobuje rozpad Cajalových tělísek, což jsou struktury v buněčném jádře spojené s pre-mRNA sestřihem, jejichž přítomnost či nepřítomnost indikuje změny v pre-mRNA metabolismu. I když buňky mutovaný protein rozpoznávají a degradují, je ho v buňkách přítomno stále dostatečné množství, které negativně ovlivňuje buněčný růst a dělení. Zjistili jsme, že buňky lze zachránit, pokud se do nich vpraví větší množství hPrp6. Tato skutečnost podporuje naši teorii, že mutovaný hPrp31 vychytává a inaktivuje funkční protein hPrp6. Tím je naznačen i nový směr výzkumu, který by – pokud bude fungovat také u lidí – mohl vést ke zlepšení stavu pacientů.
Pokud by se našel způsob, jak u jedinců postižených mutací hPrp31 zvýšit produkci hPrp6, bylo by možné oddálit nebo dokonce zabránit postižení zraku. V současné době proto pracujeme na přípravě zvířecích modelů, na nichž chceme tento postup vyzkoušet.
DAVID STANĚK,
Ústav molekulární genetiky AV ČR, v. v. i.
Toto onemocnění, kterým trpí tři až čtyři miliony lidí na celém světě, je způsobeno mutacemi různých genů, z nichž většina hraje důležitou roli v procesu detekce světla, např. genu pro opsin.
K výzkumu mutací způsobujících retinitis pigmentosa jsme se dostali oklikou. Hlavním zájmem Oddělení biologie RNA (Ústav molekulární genetiky AV ČR, v. v. i.) je způsob, jakým se v buňce zpracovává informace uložená v DNA. Konkrétně nás zajímají úpravy na úrovni pre-mRNA, tzv. pre-mRNA sestřih a komplexy, které sestřih katalyzují. Pre-mRNA molekuly jsou přesnou kopií DNA a než mohou sloužit jako templát pro syntézu proteinu, musí z nich být odstraněno více jak 9/10 sekvencí. Zbylé úseky jsou přesně spojeny, aby z nich složená informace byla čitelná a dávala smysl tak, aby mohl být na jejím základě vyroben funkční protein. Tento fascinující proces byl objeven před 30 lety, a i když jsou jeho základní zákonitosti popsány, stále není jasné, jak buňka dokáže přesně a rychle rozpoznat sekvence, které mají být odstraněny a které mají zůstat.
Vědci ve Velké Británii před několika lety zjistili, že dědičnou retinitis pigmentosa způsobují kromě mutací genů specifických pro oko i mutace v genech, jež jsou nezbytné pro přežití každé buňky v našem těle. Největším překvapením pro nás bylo, že většina těchto esenciálních genů kóduje bílkoviny důležité pro pre-mRNA sestřih! Protože jsme tyto sestřihové bílkoviny důvěrně znali a měli dobře zmapováno chování jejich nemutovaných forem, mohli jsme velmi rychle otestovat hypotézu, že mutace v první řadě ovlivňují správné formování sestřihových komplexů.
Záchrana buněk exprimujících mutovaný hPrp31 (zelený) pomocí hPrp6 (modrý). Červeně jsou označena nově vzniklá Cajalova tělíska, která indikují zlepšenou formu buněk obsahujících hPrp6.
Foto: Archiv autora
Soustředili jsme se na gen kódující protein hPrp31, který je důležitou součástí sestřihových RNA-proteinových komplexů. Zjistili jsme, že bodová mutace, která způsobuje retinitis pigmentosa, výrazně ovlivňuje chování tohoto proteinu v buňce. Zasažený protein se špatně začleňuje do sestřihových komplexů. Navíc váže – a tím inaktivuje – další bílkovinu, hPrp6, která je pro pre-mRNA důležitá. Dále jsme zjistili, že přítomnost vadného hPrp31 způsobuje rozpad Cajalových tělísek, což jsou struktury v buněčném jádře spojené s pre-mRNA sestřihem, jejichž přítomnost či nepřítomnost indikuje změny v pre-mRNA metabolismu. I když buňky mutovaný protein rozpoznávají a degradují, je ho v buňkách přítomno stále dostatečné množství, které negativně ovlivňuje buněčný růst a dělení. Zjistili jsme, že buňky lze zachránit, pokud se do nich vpraví větší množství hPrp6. Tato skutečnost podporuje naši teorii, že mutovaný hPrp31 vychytává a inaktivuje funkční protein hPrp6. Tím je naznačen i nový směr výzkumu, který by – pokud bude fungovat také u lidí – mohl vést ke zlepšení stavu pacientů.
Pokud by se našel způsob, jak u jedinců postižených mutací hPrp31 zvýšit produkci hPrp6, bylo by možné oddálit nebo dokonce zabránit postižení zraku. V současné době proto pracujeme na přípravě zvířecích modelů, na nichž chceme tento postup vyzkoušet.
DAVID STANĚK,
Ústav molekulární genetiky AV ČR, v. v. i.

 English
English
