Projekty
|
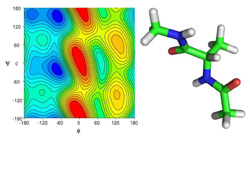
Molecular modeling and simulations of peptides
Despite of being shorter and seemingly simpler than proteins peptides manifest surprisingly complex behavior. The correct understanding and description of peptides,
their conformers and conformation preferences is difficult task both for experimentalists and theoreticians.
|
|
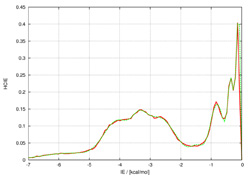
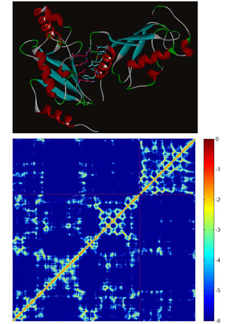
Bioinformatic analysis of protein stabilityInteractions between amino acid residues composing a protein are the major forces determining the realization of a particular protein fold and their study is a key importance towards construction of energy function differentiating between native protein state and a decoy. We perform a comprehensive study of inter-residual non covalent interactions in proteins exploiting all the available high resolution structural data. We develop a method for construction of contact maps based on interaction energy instead of geometry criteria. The final purpose of this project is a web application providing Interaction Energy Matrix analysis and energy contact maps as an analytical tool for protein structures. Further utilization of this strategy lies in protein design and mutation analysis of existing proteins. |

|
Localization hydrophobic core of globular proteinsIn this project we study a connection between non-covalent interactions of amino acids inside a hydrophobic core of a globular protein and protein stability. We develop a method for identification of hydrophobic core residues which is defined as a spatial cluster of amino acid side chains strongly stabilized mostly by vdW interactions. To quantify the stabilization of the core we use non-covalent interaction energy strength as a measure. The aim of the project is a method for redesigning of the protein hydrophobic core towards higher stability constructs. In collaboration with experimentalists working on recombinant protein expression and purification we are able to verify importance of various stabilizing side-chain interactions within the core and we can use such results for on-line predictor of more stable proteins. |
|
Protein protein interactions - a bioinformatics and computational chemistry approachProtein-protein interaction is an essential event that stays behind most of the known biological processes in a living cell. Proteins are fascinating with their ability to mutually interact in a wide range of specificity and strength. To understand the physical principles of molecular recognition and apply them in various biomedical and biotechnology branches is the major motivation underlying the research in this field. The understanding of the protein-protein interaction would also allows to influence and control processes in a cell and makes possible to design functional macromolecules and supramolecular structures. We see the path to this understanding in profound bioinformatic analysis of high-resolution crystalographic structures of protein complexes. This analysis should reveal crucial structural and statistical features of protein-protein interfaces. Computation of interaction energies between interacting amino acid side chains helps us to identify driving forces of the process. A special attention is paid to the hydrophobic residues which play important role during protein protein interaction and their hydration properties. The process of protein protein interaction can also be compared to the process of protein folding by means of the interaction properties of the composing amino acids side chains. |