Projekty
|
AntiviralsSystematic SAR studies are performed to identify novel types of antivirals, in particular compounds active against the resistant mutants that are insensitive to commonly used drugs. Activity of acyclic nucleoside phosphonates (ANPs), where the nucleobase is attached to a phosphonate moiety via a suitable linker, is based on structural similarity with the naturally occurring metabolites. ANPs are excellent lead structures for drug design because of high flexibility, absence of a labile glycosidic bond, and enzymatic and chemical stability of the phosphonate moiety compared with the phosphate ester group. ANPs can be transformed into prodrugs to improve their pharmacological properties and to increase their ability to cross cell membranes and/or target the drug to specific tissue. Effects of various structural alterations of the nucleobases and the acyclic moieties on biological activity are studied. The investigation of broad spectrum antiviral activities is performed in collaboration with Rega Institute, Katholic University Leuven (Belgium) and with Gilead Sciences (USA). |

|
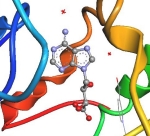
Acyclic nucleoside phosphonates with antiparasitic activityHypoxanthine-guanine-(xanthine) phosphoribosyltransferase (HG(X)PRT) is a key enzyme crucial for the survival of the malarial parasites Plasmodium falciparum (Pf) and P. vivax (Pv). Humans possess two metabolic pathways, de novo and salvage, and hence are less reliant on the purine salvage pathway for their survival. This provides a rationale for the design of inhibitors of plasmodial HG(X)PRT, which would arrest parasitemia and, at the same time, would not be toxic to the human host. Recently, a novel group of aza-ANPs has been designed, synthesized, and studied. The low Ki values of some of these compounds for PfHGXPRT and PvHGPRT and the high selectivity in favor of these enzymes compared with human HGPRT, suggest them to be lead compounds for development as antimalarial drugs. The investigation of the inhibitory activities of ANPs is performed in collaboration with the laboratory of Dr. Luke W. Guddat, University of Queensland, Brisbane (Australia). |
|
The successful development of ANP-based inhibitors of Plasmodial HG(X)PRT could inspire further research of similar drug targets in other types of parasites. The features of purine salvage system of causative organism of sleeping sickness, Trypanosoma brucei, argue for the extension of the project and therefore we have establish the collaboration with the Laboratory of Functional Genomics of Protists (Dr. A. Zíková, Biology Center v.v.i., Institute of Parasitology, Czech Republic). The extensive SAR-study of ANPs and their prodrugs against cultured cells of both life stages (procyclic and bloodstream) of T. brucei is planned, as well as experiments with the bloodstream stage with knock down of HGPRT and XPRT or APRT genes or, by contrast, experiments with overexpression of these enzymes. |
|
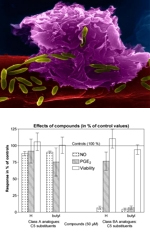
Immunomodulating agents2,4-Diamino-6-hydroxypyrimidine (DAHP) is an inhibitor of GTP cyclohydroxylase I, a key enzyme in the synthesis of tetrahydrobiopterin (BH4), which is a known cofactor of inducible NO synthase (iNOS). Several groups of DAHP derivatives with various substituents at C-2, C-4, C-5 and C-6 positions of the pyrimidine ring were prepared , which more or less effectively reduce NO and prostaglandin E2 (PGE2) production. The most effective are pyrimidines containing 2-(N,N-dimethylamino)methyleneamino group or 2-formamido group. The dual effect makes such compounds suitable mainly for the treatment of diseases which are induced, or the severity of which is potentiated, by overproduction of NO and/or PGE2. Such diseases are mainly, but not exclusively, inflammatory and cancer diseases. Our study, in collaboration with the Institute of Experimental Medicine (Dr. Z. Zídek, Academy of Sciences of the Czech Republic, Prague), is focused on the structure-activity relationship (SAR) studies of the polysubstituted pyrimidines. We hope to select several best candidates from the large portfolio of the modified pyrimidines (about 150 compounds) with potential to effectively reduce NO and PGE2 production to initiate pre-clinical development. |

|
Inhibitors of adenylate cyclaseAnthrax toxin is a three-protein exotoxin secreted by virulent strains of Bacillus anthracis - the causative agent of anthrax. Anthrax toxin is composed of protective antigen (PA), edema factor (EF), and lethal factor (LF). EF acts as a Ca2+ and calmodulin dependent adenylate cyclase (AC) that greatly increases the level of cAMP in the cell. It was found out that PMEA diphosphate (PMEApp) is able to inhibit AC from AC toxin (ACT) of Bordetella pertussis or EF of Bacillus anthracis. PMEApp, as an analogue of the natural substrate (ATP) competes for the active site of the enzyme. Synthesis of various structural analogues of PMEA as potential AC inhibitors is in progress. We have also established collaboration with the Centre for biological defence in Těchonín to study antibiotic properties of various types of compounds, but especially of properly modified ANPs. Prepared compounds will be tested against a number of pathogens (Yersinia pestis, Escherichia coli, Bacillus subtilis, Listeria monocytogenes, Salmonella enteritidis, Pseudomonas aeruginosa, Acinetobacter baumannii, Mycobacterium tuberculosis, Staphylococcus aureus and Francisella tularensis), but a special interest is devoted to bacteria causing whooping cough (Bordetella pertussis) and anthrax (Bacillus anthracis). |
| We cordially invite talented and hard-working organic chemistry students interested in medicinal chemistry to join our group and to participate in our projects. | |