
Research
We gratefully acknowledge generous financial support fom the following sources:
The J.E. Purkyně Fellowship of the Academy of Sciences of the Czech Republic
The Czech Science Foundation (GAČR) under project for the project GA15-19672S Force and Conductance in Molecular Junctions (PI: Héctor Vázquez).
The Marie Skłodowska-Curie postdoctoral actions.
About Héctor: My research focuses on the first-principles calculation of the electronic and transport properties of molecular junctions. I have also worked on the energy level alignment at organic semiconductor interfaces. Recently, I started working on modeling organic photovoltaics and exciton dissociation.
Single molecule transport
I started working on single molecule transport during my postdoctoral stay at DTU Nanotech, with Antti-Pekka Jauho and Mads Brandbyge. There I started using the TranSIESTA code. I later joined the group of Latha Venkataraman, where I was carrying out the theretical work co-supervised by Mark S. Hybertsen.
Constructive quantum interference and the conductance superposition law
This project analyzed the conductance superposition law in molecular circuits by comparing the conductance of molecules that have two backbones bound in parallel to that of their single-backbone counterparts. In other words, for a molecule that has two backbones bound to the electrodes in parallel, is the total conductance the sum of the conductance of each backbone?
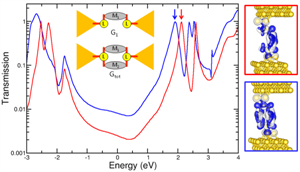
Through the combination of chemical synthesis, simulation and experiment, we found that the conductance of double-backbone molecues can be more than twice (eg. ~3×) that of the single-backbone counterparts due to quantum interference: the scattering states propagate coherently through both molecular backbones in parallel. First-principles calculations show that the conductance increase between the double- and single-backbone molecules is due to quantum interference (Nature Nanotechnology News & Views).
Highly-conducting metal-molecule links
This project demonstrated highly-conducting contacts for single-molecule transport. These metal-molecule contacts are covalent bnds between Au atoms and the terminal C atoms in the molecule. The formation of these bonds comes from molecules initially synthesized with trimethyl tin (SnMe3) linker groups. The SnMe3 groups became detached in-situ at the junction as the transport experiments were carried out, resulting in the formation of direct, covalent Au-C bonds to the molecule. For alkanes, the measured conductance is almost 100 times higher than with any other termination measured so far.
The high conductance of Au-C bonds results from a strong electronic coupling, with a broad transmission resonance slightly below the Fermi level. Calculations can probe the intrinsic limit of the linker only throgh simulations of transmission through just one methyl unit. The calculated spectrum shows a very high transmission and almost ideal contact resistance over a wide energy range, making Au-C bonds excellent linkers for single molecule transport.


We then extended our study to the case of conjugated molecules, where the highly conducting Au-C links were fully exploited by coupling them to a molecular π system. Experiments and calculations yielded almost unit transmission across a conjugated molecule having one phenyl unit, where the junction has a ~8Å gap.

Tuning and guiding current flow in tin-phthalocyanine molecules
The contacts between the molecule and the electrodes are important. In this project, the degree of coupling between a SnPc molecule and the Ag(111) substrate was tuned by selectively forming chemical bonds between any or all four molecular ligands and the substrate. Through selective dehydrogenation, covalent bonds between one, two, three or all four SnPc ligands and the substrate were formed. Transport calculations were able to explain the seemingly paradoxical behavior measured experimentally, where increased contacts to the substrate result in a conductance drop. Calculations showed that, despite the stronger bonding to the substrate when more ligands were bound to the metal substrate, the molecular π system, responsible for transport, was disrupted and pushed away from the Fermi level. Additionally, the calculations provided a picture of quantum local (atom-atom) currents and how these change when covalent bonds to the metal substrate were formed.

Organic photovoltaics in model Donor/Acceptor Hamiltonians
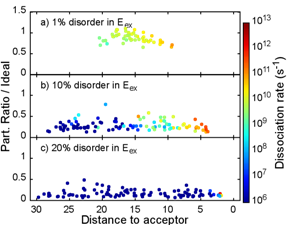
In 2012, I was a postdoc in the group of Alessandro Troisi at the University of Warwick. I worked on the theory of organic photovoltaics focusing on exciton dissociation in model Donor/Acceptor systems. How efficient organic photovoltaic cells generate free charges from incident light has been studied extensively but is not yet well understood: in order to separate, electron and hole have to overcome their Coulomb attraction, which is rather strong in such low dielectric constat materials. Several mechanisms have been proposed but no consensus exists. Recently, “hot” excitons having excess energy have received attention since they have been shown to result in efficient exciton dissociation. We presented a method for calculating the rates of exciton dissociation in model Donor/Acceptor interfaces. The model describes clearly and compactly the process of charge generation and the role of disorder on exciton dissociation rates.
Initial states are Frenkel excitons, while final states are Charge-Transfer (CT) states. Using a widely applicable Hamiltonian and a Green’s function approach, we calculated the generation rates of CT states. We focused on the role of disorder and of the delocalization of the excitonic wavefunctions. Disorder in the initial states results in a clear exponential dependence of dissociation rate with respect to the distance to the interface, with an exponential decay constant β that increases linearly with the amount of disorder. Disorder in the parameters describing final (CT) states has little effect on the dissociation rates. In all cases, the excitons dissociate into “hot” CT states with partially separated charges, where electron and hole are located far from the interface.

Energy level alignment at organic semiconductor interfaces
My PhD thesis, ‘Energy Level Alignment at Organic Semiconductor Interfaces’ was carried out at the Universidad Autónoma de Madrid and supervised by Fernando Flores. My work dealt with the theoretical description of the energy level alignment and electronic structure at interfaces of organic semiconductors. While the relevant energy levels of the isolated systems can be calculated or measured, the offsets once the interfaces are formed depend on the electronic structure of the junction. My PhD proposed a model based on the Charge Neutrality Level to understand and predict energy level alignment at metal/organic and organic/organic interfaces. Our results showed that level alignment could be described by the tendency of the CNL of the organic material to align with the metal work function, or with the CNL of the other organic material in the case of organic heterojunctions. The CNL thus plays the role of an effective electronegativity of the organic material.

Having to model energy level alignment at organic interfaces presented the problem of DFT self-interaction errors. These result in band line-up errors and are related to the well-known underestimation of the gap by DFT. My PhD developed a model, based on a simplified treatment of a many-body Hamiltonian, for correcting the DFT level positions through orbital-dependent shifts to the molecular levels. These self-interaction corrections improve on the DFT molecular level positions and on the gap problem, in agreement with accurate many-body calculations. This simplified self-interaction correction scheme captures the most important many-body contributions to the total energy not accounted for in DFT.